CoNVaQ
CoNVaQ is a web tool for CNV-based association study between two groups of samples. The simple web interface allows you to quickly upload segmented CNV samples and search for variations that are overrepresented in a population. CoNVaQ provides two models for determining significance of genomic regions:
- A statistical model using Fisher’s exact test.
- A novel query-based model allowing you to specify which CNVRs are considered significant using simple queries, e.g. “find the largest region duplicated in ≥ 30% of cases and ≤ 5% of controls”.
Furthermore, CoNVaQ computes empirical p-values (called q-values) for matching regions by repeatedly executing the same query while randomly perturbing the sample populations. The q-value is the probability of finding a matching region of same length or longer, when the size of each group is preserved, but the samples are randomly distributed among groups. The position and type of the individual CNV segments are not changed by the perturbation.
For more information on how to use CoNVaQ, see our step-by-step guide or watch the screencast to the right.
Screencast

R package
CoNVaQ is also available as an R package. You can install the latest development version using devtools with:
devtools::install_github("SimonLarsen/convaq")
Package documentation:
- Vignette: Short introduction
- Reference manual
Citation
If you use CoNVaQ in your research, we kindly ask you to cite the following publication:
Larsen SJ, do Canto LM, Rogatto SR, Baumbach J, 2018. “CoNVaQ: a web tool for copy number variation-based association studies”. BMC Genomics. Springer Nature.
Upload data
Select the species and genome assembly of interest, then upload sample files for each group. For information on how to structure the data, see the synthetic example data set and the file format specification.
Please wait for files to finish uploading before hitting Submit.
Analysis
Data set summary
Model
Choose the model to use below.
Merging regions
Compute empirical p-values through permutation tests.
Results
Results
- Click on the icon to show detailed information about a region.
- Select rows for analysis by clicking on them. Click on a row again to deselect it.
Export results
Download all regions, including variant frequencies and individual patient states:
Download as CSV Downloads as Excel Download PDF reportGuide
Contents
Segment file format
Segmented CNV calls must be uploaded to CoNVaQ in the form of a data table. Two files must be uploaded: one corresponding to each group of patients/samples. CoNVaQ supports uploading plain-text files (such as CSV, TSV) as well as Microsoft Excel files. Each segment file must have the following format:
- Each row must have five columns.
- The first row is mandatory and contains the file header.
- Each following line (row) describes a segment.
- The order of the columns matter and should match the description below.
The content of each column is described below.
chr prefix.
Gain, Loss or LOH (lack of heterozygosity).
Example
Below is an example file containing two patients with two variations each, and a third sample with no variations.
| name | chr | start | end | type |
|---|---|---|---|---|
| PAT1 | 1 | 53131738 | 109684893 | Gain |
| PAT1 | 1 | 174832716 | 179354603 | Gain |
| PAT2 | 18 | 38525729 | 38541321 | Loss |
| PAT2 | X | 104713140 | 104715491 | Loss |
A synthetic example data set is available here.
Step-by-step guide
This is a step-by-step guide on how to use CoNVaQ. It will guide you through the process of performing a basic association study using the two supported models.
Uploading files
Before starting your analysis, you must upload the files containing your segmented CNV calls. First, select the species and genome reference your CNV calls were generated from. If your species is not found in the list, select Other instead. Some functionality will be disabled when no known species is selected. Here we select species Homo sapiens and reference genome NCBI36/h18.

Next you must upload the CNV calls for your two groups of samples. You can either upload a single file for each group or multiple files (i.e. one file per patient). See the file format specification for more information on how to format the input files. You can also choose to give each group a descriptive name to make them easier to tell apart in the analysis. Here, we name the two groups HPV-positive and HPV-negative.

Once you press Upload data the files are uploaded and you are taken to the analysis page. On the top of the page you will find a table summarizing the data set. In this example we observe that the HPV-positive group contains 14 samples while the HPV-negative group contains 27.

Statistical model
First we want to search for significant CNV regions using the statistical model. In order to use the statistical model we first need to select the Statistical model tab below the summary table.
Under Options you can set the parameters for the statistical test. Here we choose to use a one-sided test with a p-value cutoff of 0.05. We check merge adjacent regions and use a value of 0, meaning adjacent CNV regions are only merged if they are directly adjacent. Finally, press Submit to search for significant CNV regions.

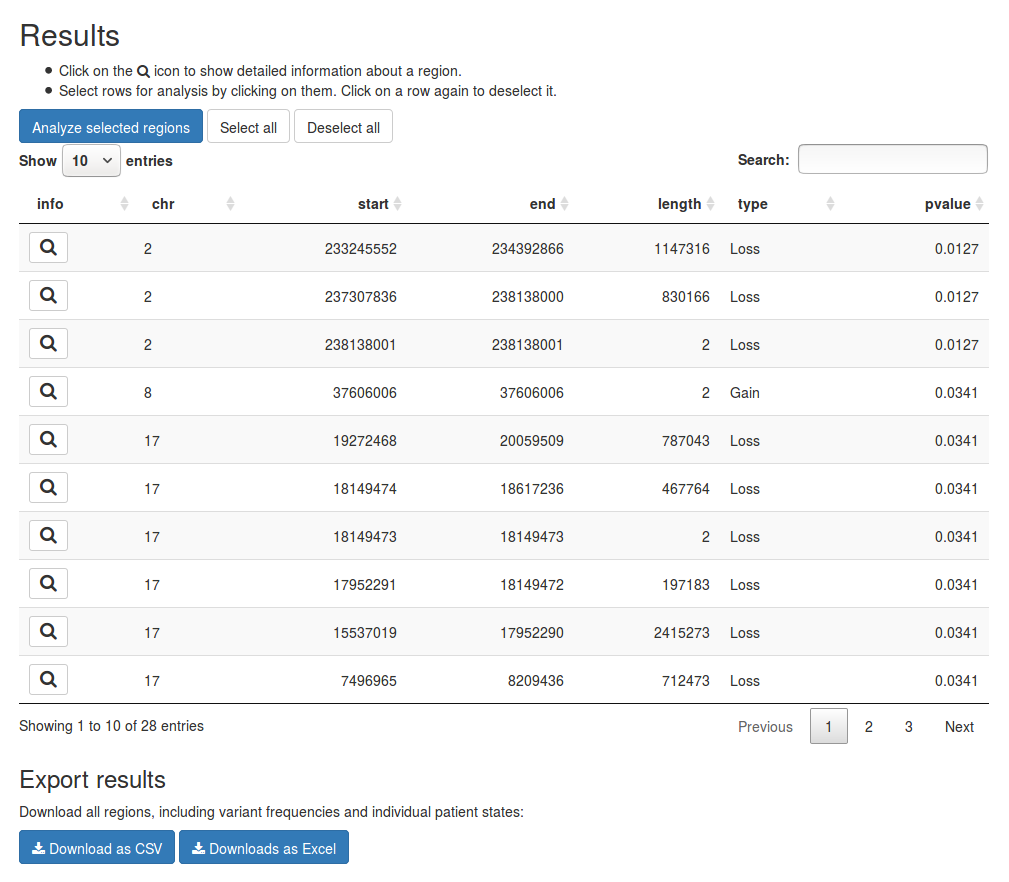
The results of the analysis will be presented in the results table under Results. Each row represents a genomic region that was deemed significant. The first seven columns contain the location and size of the region, the type of event detected and the p- and q-values if applicable. The remaining six columns give the frequency of CNVs in the region for each group.
In this example we observe that in the first region 21% of samples in the HPV-positive group have a loss of copy numbers while all samples in the HPV-negative group have normal copy numbers. We also observe that 0%-7% of samples in the HPV-positive group have a gain in copy numbers. When a frequency is represented as a range it means the region was created by merging two or more regions. The range then represents the smallest and greatest frequency within the region.

If you wish to export the data in the results table you can use the buttons below the table. They allow you to save the results as either a plain-text CSV file or an Excel table.

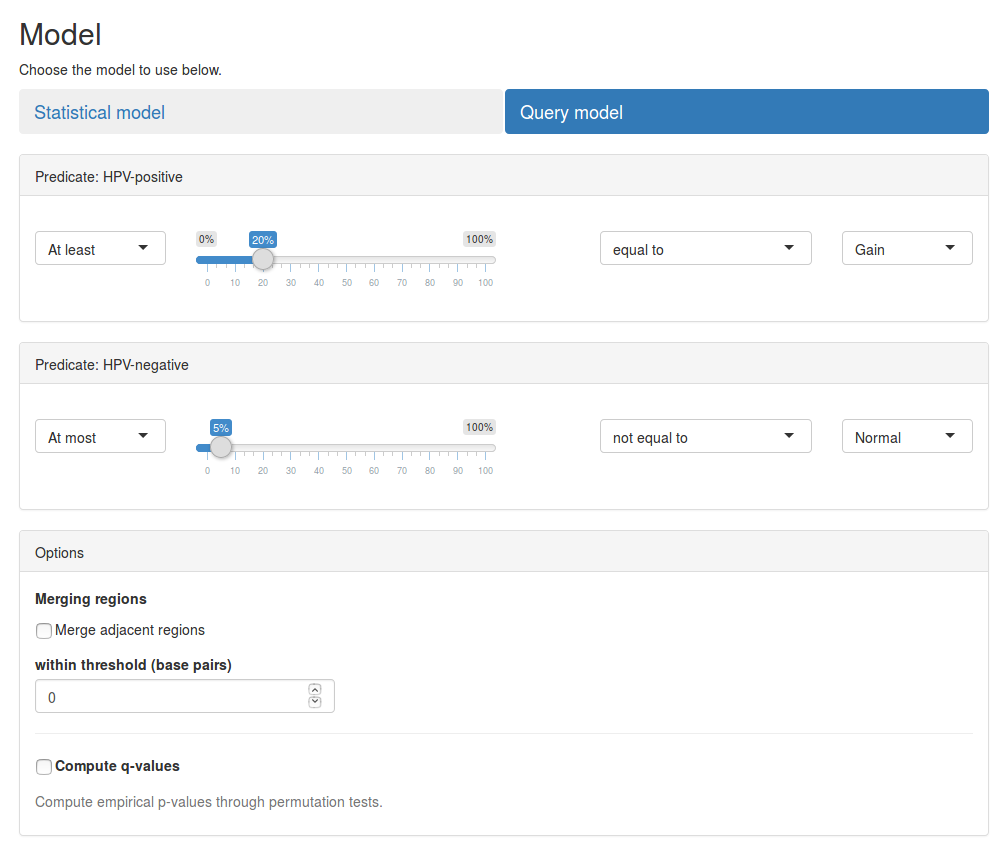
Query model
Next we want to search for significant CNV regions using the query-based model. Select the Query-based model tab below the summary table to get started.
We will use the following query to search for loss events:
Q = ((≥, 20%, =, Loss), (≥, 100%, =, Normal))
This query specifies, that we are searching for regions where at least 20% of samples in the HPV-positive have a loss in copy numbers and all samples in the HPV-negative group must have normal copy numbers (see the image below). See our paper for a description of how queries are defined and evaluated.

Press the Submit button to search for regions matching this query. The results will be shown in the results table below. The results table for the query-based model contains the same information as for the statistical model, except that p-values are not computed.

Inspecting CNV regions
You can inspect the individual reported CNV regions by clicking on the next to them. This will bring up the inspection window.

Under Summary you can find the statistics for the region also presented in the results table. Under Sample states you can obtain the state of each sample. Click on the name of a group to expand the panel.
In our example, if we expand the HPV-positive group, we can see that a loss of copy numbers was observed in patients 1389 and PE11T, among others.

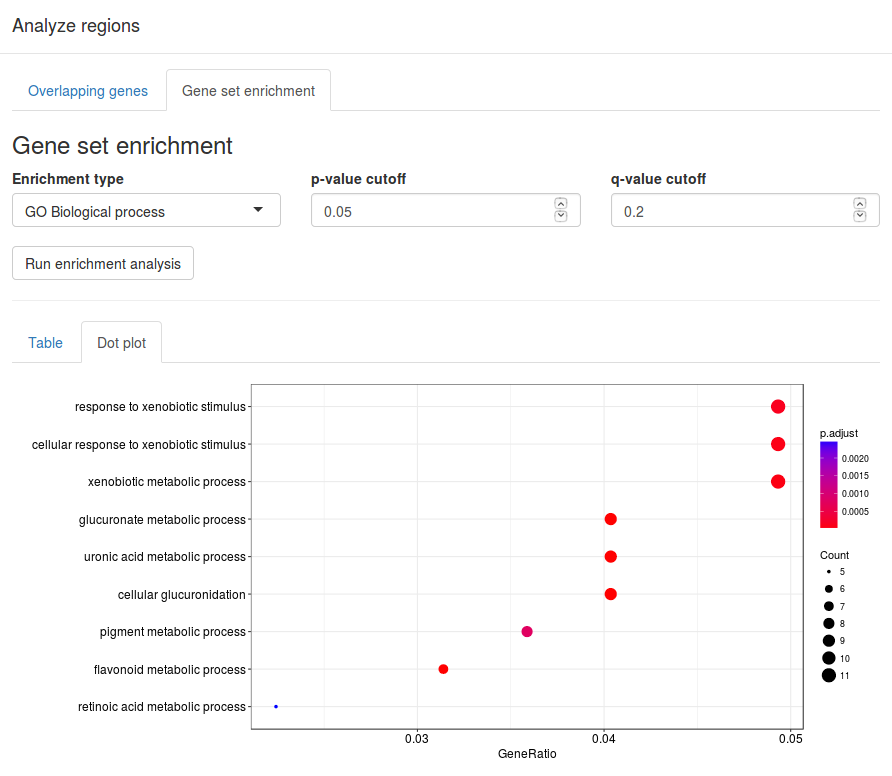
Enrichment analysis
After identifying a set of CNV regions one can perform a simple gene set enrichment analysis. In the results table, select one or more regions by clicking on them. Selected regions will be highlighted with blue. Click on a region again to deselect it.
After selecting the regions of interest click the Analyze selected regions button above the results table. This will bring up the analysis window.

In the analysis window you will find a table containing all known genes overlapping one of the previously selected regions. To perform gene set enrichment on the set of overlapping genes, select the Enrichment analysis tab on the top of the window. Under Enrichment type select the type of annotations to search for, then click Search to perform the analysis. The results will appear in a table below, showing each matching annotation, the p-value for overrepresentation and a list of matching genes.

In this example we select Reactome pathways in order to search for overrepresented Panther pathways. We use a p-value cutoff of 0.05 and a q-value cutoff of 0.2.


About
Contact
If you want to contact us regarding CoNVaQ, please contact us in the following order:
Source code
The source code for CoNVaQ as well as the web frontend is available under an MIT license here:
CoNVaQ web version 0.1.0